Z. Yang, S. P. Pujari, R. Armstrong, K. Mathwig, F. Leonardi, M. M. J. Smulders and H. Zuilhof, Langmuir (2026), published online. [link]
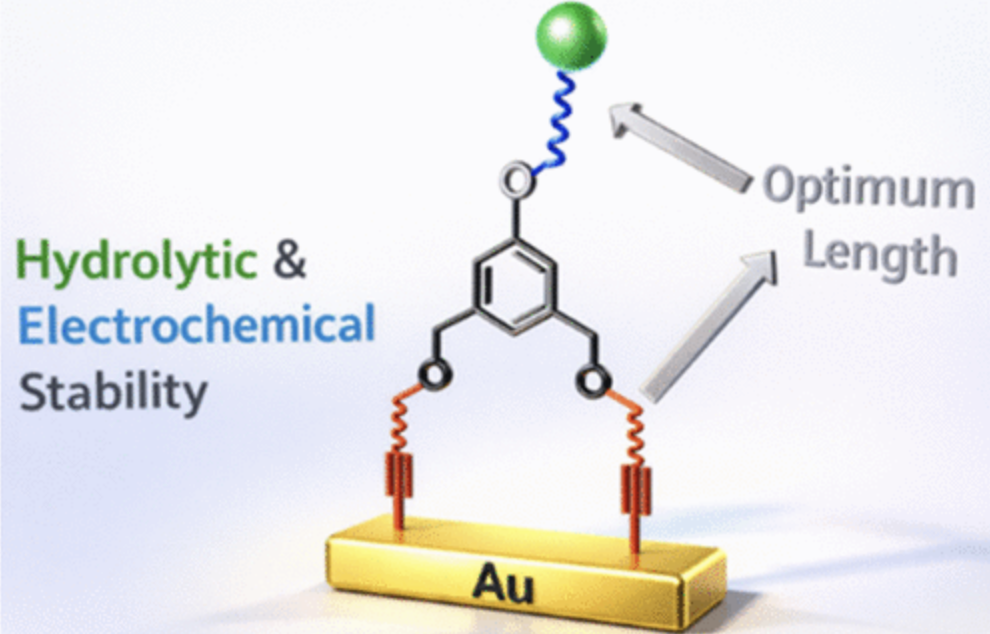
The terminal alkyne-gold bond provides strong self-assembled monolayers (SAMs) with significant potential in biosensing applications. However, to deliver on this potential, a deeper understanding of their electrochemical stability in relation to their molecular structure is required. This study systematically investigates the electrochemical stability of a series of bidentate alkyne monolayers on gold (Au) (CnH2n+1O–C6H3–[meta-CH2O–(CH2)mC≡C–Au]2; head: n = 4, 10, 18; foot: m = 1, 3, 9) focusing on the effects of different head chains and the less-explored ‘two-legged’ foot chain structures on the surface coverage (θ) and interfacial charge transfer resistance (Rct). θ remains nearly constant with increasing potential up to 0.9 V vs Ag/AgCl (3 M KCl), followed by a sharp drop between 0.9 and 1.0 V. In contrast, the corresponding Rct decreases over a wider voltage range, with different monolayers showing distinct profiles and no distinct “onset” potentials. Overall, monolayers with long foot chains exhibit the highest electrochemical stability. Furthermore, we prepared two −CF3-labeled monolayers and evaluated their hydrolytic stability in different aqueous media by monitoring changes in the F 1s/Au 4f ratio by XPS. The results confirm that long foot lengths (m ≥ 3) also provide good hydrolytic stability. These structure–functionality findings provide further impetus for their further application in sensor interface engineering.